(I). Overview
1. Medical Devices (Non in vitro diagnostic reagents, mainly refers to passive medical devices, active medical devices)
According to the Regulations on Supervision and Administration of Medical Devices (State Council Decree No.680), the category I of medical devices shall be subject to product filing management, and the categories II and III of medical devices shall be subject to product registration management.
When the categories of medical devices cannot be determined, the following methods can be adopted:
• Inquire according to the Rules for the Classification of Medical Devices, Category and Classification List of Medical Devices or the same products already on the market.
• Apply directly according to the category III of medical devices
• Apply for classification definition
• Apply for attributes definition for the combined products of drugs and devices
2. In Vitro Diagnostic Reagents
According to the Measures for the Administration of Registration of In-Vitro Diagnostic Reagents, the category I of in vitro diagnostic reagents shall be subject to filing management, and the categories II and III of in vitro diagnostic reagents shall be subject to registration management.
When the categories of in vitro diagnostic reagents cannot be determined, the following methods can be adopted:
• Inquire according to the classification requirements in the Measures for the Administration of Registration of In-Vitro Diagnostic Reagents or the same products already on the market.
• Apply directly according to the category III of in vitro diagnostic reagents
Apply for classification definition
(II) Filing
1. Filing Basis
According to the Regulations on Supervision and Administration of Medical Devices (State Council Decree No.680), the category I of medical devices shall be subject to product filing management. According to the Measures for the Administration of Registration of Medical Devices, the filing of medical devices refers to the process that the filing applicant of medical devices submits the filing data to the Food and Drug Administration who will then archive the submitted data for future reference.
According to the Measures for the Administration of Registration of In-Vitro Diagnostic Reagents, the category I of in vitro diagnostic reagents shall be subject to filing management.
2. Qualification Requirements for Filing Application
Imported medical devices/in vitro diagnostic reagents for filing shall be approved for sale in the country (region) where the filing applicant's place of registration or production is located.
If the country (region) where the filing applicant's place of registration or production is located does not manage the product as a medical device, the filing applicant needs to provide relevant certification documents, including the certification documents that the country (region) where the applicant's place of registration or production is located allows the product to be marketed.
3. Filing Procedure
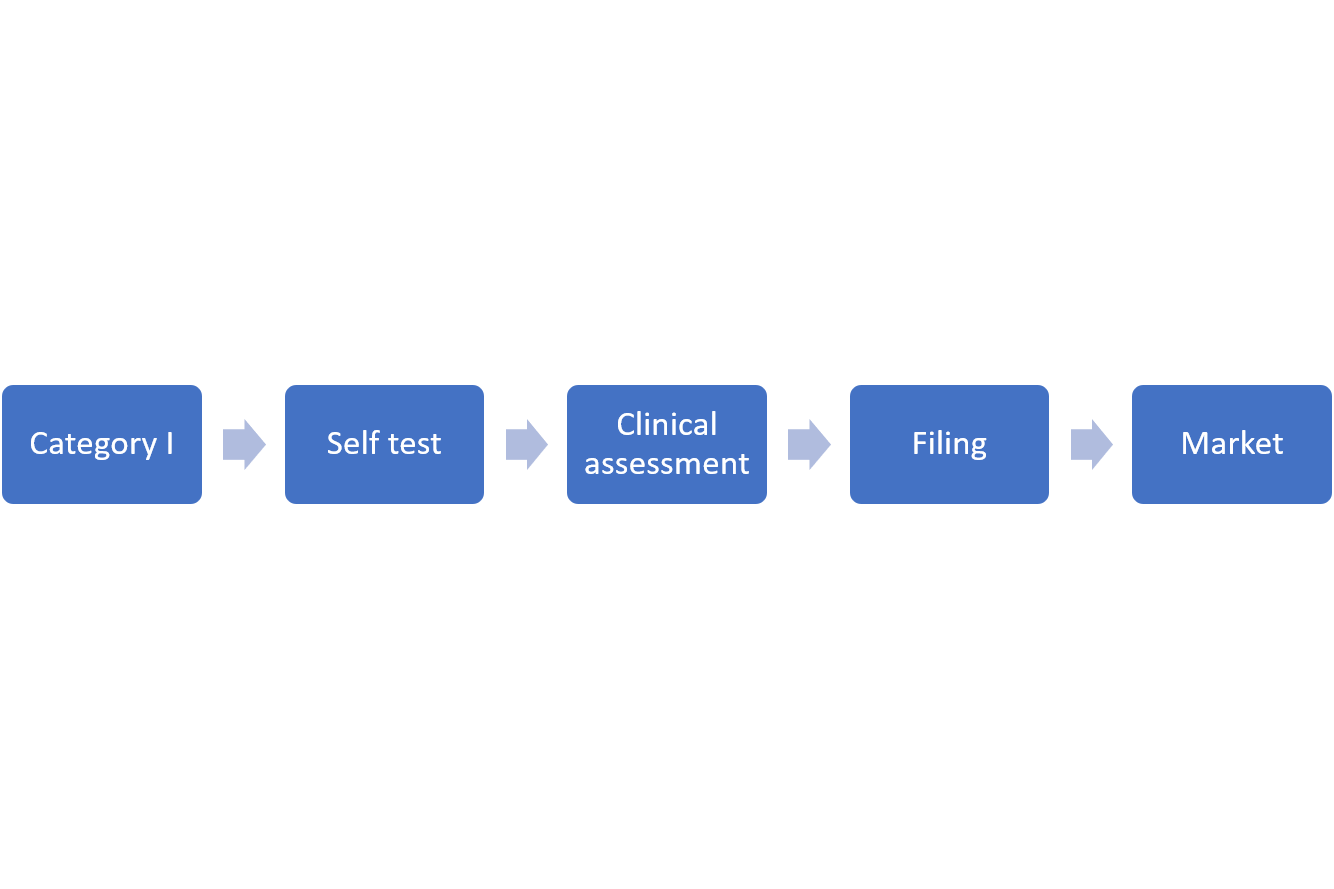
4. Material Requirements for Filing Application
1) The following materials shall be submitted for the filing of Category I medical device products:
| File Name | Category I |
| 1. Filing Form | √ |
| 2. Certification documents | √ |
| 3. Production and manufacturing information | √ |
| 4. Clinical evaluation data (Category I filing does not require clinical trials) | √ |
| 5. Product risk analysis data | √ |
| 6. Technical requirements of product | √ |
| 7. Product test report | √ |
| 8. Product manual and label samples | √ |
| 9. Declaration of conformity (the medical device filing applicant shall be responsible for the authenticity of the submitted materials) | √ |
2) The following materials shall be submitted for the filing of Category I in vitro diagnostic reagent products:
| File Name | Category I |
| 1. Filing Form | √ |
| 2. Certification documents | √ |
| 3. Production and self-inspection record (production and manufacturing information) | √ |
| 4. Clinical evaluation data (Category I filing does not require clinical trials) | √ |
| 5. Product risk analysis data | √ |
| 6. Technical requirements of product | √ |
| 7. Product registration test report | √ |
| 8. Product manual | √ |
| 9. Label samples | √ |
| 10. Declaration of conformity | √ |
(III) Registration
1. Registration Basis
According to the Regulations on Supervision and Administration of Medical Devices (State Council Decree No.680), Category II and Category III medical devices shall be subject to product registration management.
As per the Measures for the Administration of Registration of Medical Devices, registration of medical devices is a process in which the Food and Drug Administration performs a systematic evaluation over the safety, effectiveness study and its results of the medical devices to be marketed as per the application filed by the medical device registration applicant and legal procedures to decide whether to approve the application.
Category II and Category III in-vitro diagnostic reagents shall be subject to registration management pursuant to the Measures for the Administration of Registration of In-Vitro Diagnostic Reagents.
2. Qualification Requirements for Registration Application
The imported medical devices subject to registration application shall be approved for marketing in the filing applicant's place of registration or the country
(region) where the production site is located.
Provided that the product is not included in medical devices for management in the filing applicant's place of registration or the country (region) where the production site is located, the filing applicant is requested to submit relevant certification documents, including the certification documents proving that the product is permitted to be marketed and sold in the place of registration or the country (region) where the production site is located.
3. Registration Procdure
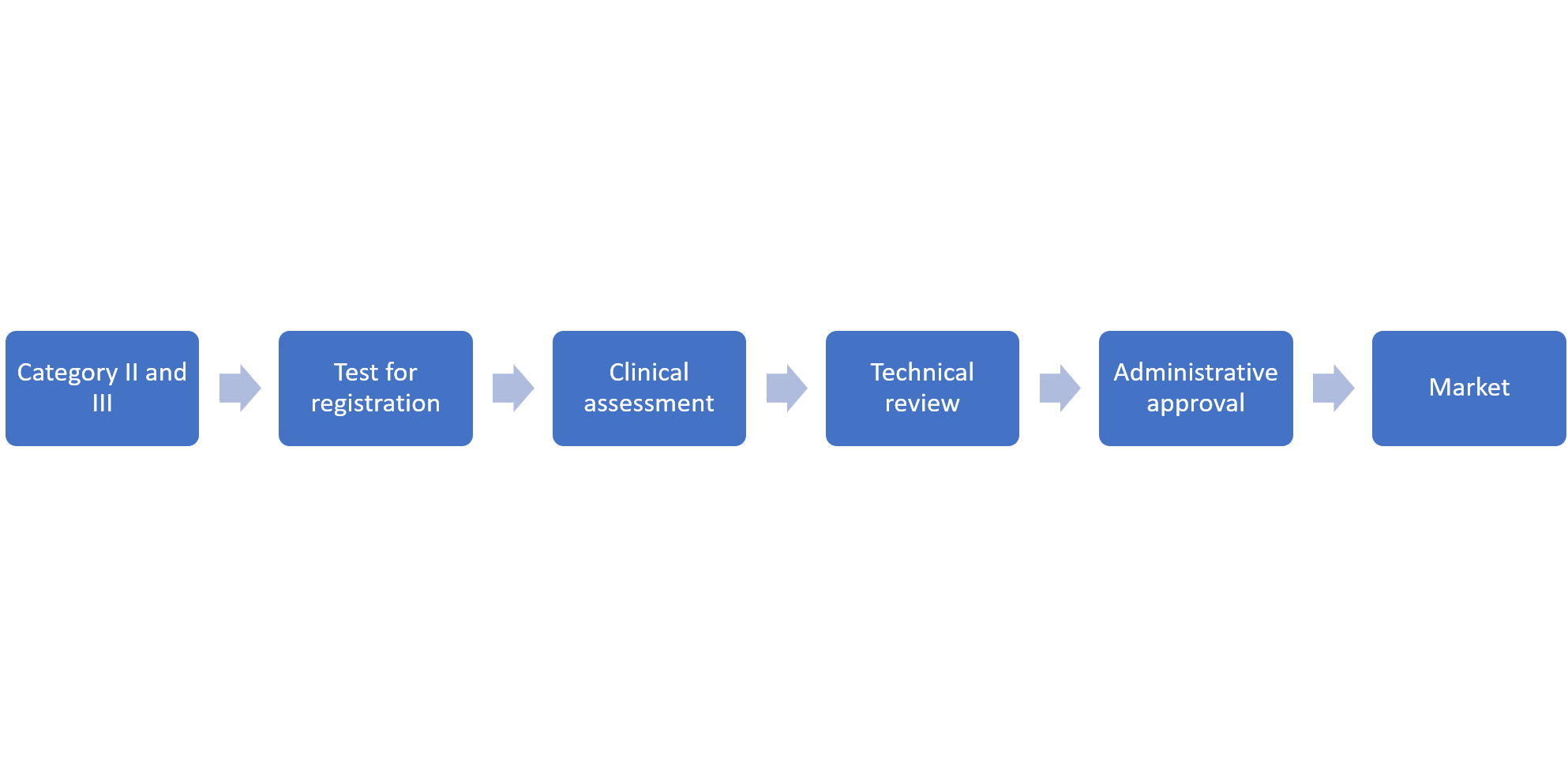
4. Material Requirements for Registration Application
1) The following data shall be submitted for registration of Category II and Category III medical device products:
| File Name | Category II | Category III |
| 1. Application form | √ | √ |
| 2. Certification documents | √ | √ |
| 3. List of basic requirements for safety and effectiveness of medical devices | √ | √ |
| 4. Summary data | √ | √ |
| 5. Study data | √ | √ |
| 6. Production and manufacturing information | √ | √ |
| 7. Clinical evaluation data | √ | √ |
| 8. Product risk analysis data | √ | √ |
| 9. Product technical requirements | √ | √ |
| 10. Product registration test report | √ | √ |
| 11. Product manual and label samples | √ | √ |
| 12. Declaration of conformity (the medical device registration applicant shall be responsible for the authenticity of the submitted data) | √ | √ |
2) The following data shall be submitted for the registration of Category II and Category III in-vitro diagnostic reagent products:
| File Name | Category II | Category III |
| 1. Application form | √ | √ |
| 2. Certification documents | √ | √ |
| 3. Summary data | √ | √ |
| 4. Study data of main raw materials | √ | √ |
| 5. Study data of main production process and reaction system | √ | √ |
| 6. Performance analysis and evaluation data | √ | √ |
| 7. Data for determination of positive result or reference range | √ | √ |
| 8. Stability Study Data | √ | √ |
| 9. Production and self-test record | √ | √ |
| 10.Clinical assessment data | √ | √ |
| 11. Product risk analysis data | √ | √ |
| 12. Technical requirements of product | √ | √ |
| 13. Production registration test report | √ | √ |
| 14. Production operation manual | √ | √ |
| 15. Label sample | √ | √ |
| 16. Declaration of conformity | √ | √ |
5. Trial Requirements
1) Medical devices of categories II and III
For application of registration of medical devices of categories II and III, clinical trial shall be carried out. However, those exempted of clinical trial shall be excluded. Clinical trial refers to the confirmation or validation process in a qualified clinical trial institution of medical devices for the safety and effectiveness of medical devices for which registration application is filed in normal operation condition.
• Clinical trial of medical devices shall be carried out in a qualified clinical trial institution in accordance with the Good Clinical Practice for Medical Devices. The production of samples for clinical trial shall comply with relevant requirements of the quality management system for medical devices.
• The sponsor shall organize to formulate clinical trial protocol in accordance with the category, risk and intended purpose etc. of the medical device.
• Small sample feasibility test shall be carried out before the design of clinical trial protocol.
• A clinical trial protocol shall include:
a) General information
b) Background of clinical trial
c) Purpose of the trial
d) Trial design
e) Safety assessment method
f) Effectiveness assessment method
2) In vitro diagnostic reagents of categories II and III
For application of registration of in vitro diagnostic reagents of categories II and III, clinical trial shall be carried out. However, those exempted of clinical trial shall be excluded. Clinical trial of in vitro diagnostic reagents (including comparison study trial for marketed product) refers to systematic study on in vitro diagnostic reagents in corresponding clinical environment.
• Applicants of category III products should select not less than 3 (including 3) and applicants of category II products should select not less than 2 (including 2) clinical trial institutes that have obtained qualification to carry out clinical trial in accordance with relevant regulations. The production of samples for clinical trial shall comply with relevant requirements of the quality management system for medical devices.
• Applicants should sign clinical trial contracts with the clinical trial institutions, formulate and improve the clinical trial protocol with reference to relevant technical guidelines and provide samples for clinical trial free of charge and assume the costs of clinical trial.
• Clinical trial institutions should issue clinical trial reports respectively after they have completed the clinical trial. The applicant or leading unit of the clinical trial should summarize the results of clinical trial and complete the summary report of the clinical trial in accordance with relevant technical guidelines.
IV. Duration
1. Import medical devices/in vitro diagnostic reagents of category I:
| Main Step | Estimated Time |
| Collect and sort filed data | As per the specific condition |
| Test | As per the specific condition (self-test report) |
| Clinical assessment data | As per the specific condition (clinical trial not required) |
Accept by acceptance institution |
If the filed data complies with the requirements, the Food and Drug Administration should file on the site; if the filed data is not complete or does not comply with the required form, all the missing contents to be supplied should be notified to the filing person in one time, and the filing person should file after supplying and correcting the data. |
| Total | Depend on the material preparation time |
2. Import medical devices/in vitro diagnostic reagents of categories II and III:
| Main Step | Estimated Time |
| Collect and sort registered data | As per the specific condition |
| Registration test | As per the specific condition |
| Clinical trial | 1-2 years |
| Accept by acceptance institution | 5 working days |
| Hand over to the review institution | 3 working days |
| Technical review | Category II: 60 working days Category III: 90 working days |
| Material correction (if necessary) |
(within)1 year |
| Technical rewiew after material correction | 60 working days |
| Administrative approval | 20 working days |
| Accreditation | 10 working days |
| Total | 2-3 years |